

#AskMeAboutPorphyria
Porphyria Awareness Week is an international campaign that many countries around the world embark upon. It is held in April each year and has a slightly different theme every year.
This year, the international porphyria community have been facing and trying to adapt to a completely new way of living, working and educating our children in this new world that we are all facing!
In the UK and Ireland, we continued our efforts to raise awareness in an adapted way (something that us porphyria patients are all well equipped for) by bringing the porphyria community together across social media platforms!
We wanted to make sure that porphyria patients felt and continue to feel less isolated and able to interact with each other.
8 Days of Porphyria
In the run up to Porphyria Awareness Week 2020, many of you shared your patient stories in our 8 Days of Porphyria. Thank you to you all for taking the time to tell us how you are affected.
Read on to hear these amazing and heartfelt stories that were so kindly shared to help support others and raise awareness.
Alicia’s AIP journey



Hello! I’m Alicia, I’m 25 years old and I have Acute Intermittent Porphyria (AIP).
Oh gosh where do I start, I currently live with my partner of 5 years, Ricky and our cat Peppa. I guess I would explain my personality as being loud, gobby and family orientated.When I was asked to write a story about myself, one of the questions I thought about was what do other people think of my personality? So I sent all my family, close friends and my work colleagues a message asking what they thought about me. The 3 main points that everyone came back with was that I am generous, always going out of my way to help others, determined and STUBBORN!
But the message I received from my younger sister, Michelle, had me in tears for 5 minutes. She said I inspired her. Now that might not seem like a lot to some people but to me it meant the world. My siblings are everything to me; Julia, Ben and Michelle are all younger than me. I’m the only person that can make them cry without getting angry; if someone else upsets them that’s it – WAR! My mum always tells the story of me being at my brother’s football matches. If Ben gets hurt or pushed that’s it – I’m the first one on that pitch picking fights with children 10 years my junior. How nasty does that make me sound, haha!
I grew up with my mum and Ben in Gillingham from the age of 9, but I was originally from Croydon. My mum and Dad spilt up when I was really young and my Dad went on to get married and have my sisters, so I guess you could say I have two families. I work for Medway Sport and I love my job; I volunteered for them for about a year, helping out at their sporting events and then I was offered an admin job off the back of that. My work colleagues are like my family at work, they are always there to support me, guide me and help me whenever they can and they are a great bunch of people and always up for a laugh.
I don’t really have many hobbies unless you class Starbucks dates and B&M shopping a hobby but what I love to do is spend time with my close friends and family. I don’t have many friends but the friends I do have are gems, they are precious and very rare.
BEFORE DIAGNOSIS
Before I got diagnosed with AIP, I was working by the age of 15 at a Fish and Chip restaurant and most weekends I was out with my school friends, going shopping before we were 18 and then partying when we reached 18. I was a very strange 18 year old because I didn’t like drinking, I didn’t like the smell or the taste, so I decided then that when I reached 17, I would learn to drive so I could safely take my friends home after a night out. I also wanted my independence, I wanted my freedom! So that’s exactly what I did, I passed within the year and my employers at the time brought me my first car, a little Ford KA. I’m still in contact with my first employers and I wouldn’t even call them employers, I would call them my Greek family. They taught me some very important life lessons and skills that I still use every day now. That pretty much sums up my life before I fell poorly.
DIAGNOSIS
Now here comes the hard bit, talking about the thing that hurts me most in this world; Acute Intermittent Porphyria. I first got diagnosed at the age of 19 on the 14th March 2014 at Medway Hospital. It all started because I caught a simple chest infection, so like everyone else I went to my doctors to ask for some antibiotics but what we didn’t know was that porphyria patients are allergic to many medications. So I started these antibiotics and within 2 days I started getting the symptoms of tummy pains, sickness, fatigue and confusion but being the stubborn person I am I continued to work and carry on with my life.
While I was at work I started having problems with my breathing and the pain in my tummy was crippling so my employers rang my mum and the ambulance and before I knew it I was getting rushed into hospital. I can’t really recall what happen in A&E or what happen for the next 10 days. According to my family, it was the most terrifying thing they’ve ever seen! I wouldn’t stop crying in pain and I was very reluctant to allow people to touch me. I wouldn’t eat, or sleep. I just wanted the pain to stop. I didn’t want to be here. I wanted it all to end, however that end may be. I was tested for cancer, for countless amounts of infection, for a hernia but they all came back clear.
Somewhere down the line it came out that my Nan, on my Dad’s side, has porphyria and whether I had been tested for it ……….. Porphyria …….. What’s porphyria? Porphyria hadn’t been heard of by Medway Hospital and it took a professor from Kings College Hospital in London to organise the testing and within 2 days I was given the correct medication to treat the porphyria attack and on the road to recovery. Once I was well enough to understand what was going on a Doctor from Kings and Medway came and spoke to me to explain what had happened and what porphyria was. I wasn’t given any kind of leaflet or booklet explaining what porphyria was, like you get when you get cancer, I got nothing and felt like… that’s it we are on our own!
It was very hard at the age of 19 to understand that I was different, that I now needed to be extra careful. I didn’t want to believe I was ill and I ignored all the symptoms and signs I was experiencing, all the pain in my body. I was told I would need to eat properly and rest more. Well I wasn’t having that – I carried on not eating properly and continued to work and party.
What was it that people were calling me at the beginning? Stubborn, maybe they are right! Well none of this worked in my favour and every single period after this first attack I continued to have recurring attacks and land myself in hospital, spending no less than 2 weeks at a time. So the decision was made to medically stop my periods to see if this would help, and it did for 9 months! Great I continued with my life, I forgot I was poorly, I was normal again.
After 9 months my body became immune to the medication and I had another attack. So I made the decision to slow down a little. I continued my work but stopped going out every weekend. This helped, but not by much. Sadly, it took a life threating attack, complete paralysis and 6 months in two hospitals for me to realise that actually I am different, I am really poorly and if I don’t listen I could die. This put an emotional strain on my relationship with my partner, my family and my friends. I had to learn to walk again because the porphyria had caused an excessive amount of nerve damage to my legs. Everything I touched was hurting, the sensations felt really weird. I didn’t like people touching me. So after this attack I stopped work all together as I wanted to concentrate on my recovery and maintain my relationships with people. I took a whole year out of work but mentally I was struggling. I’m not that sit at home girl. So I made the decision to volunteer and that’s how I got my part time job at Medway Sport.
MY LIFE NOW & MY FUTURE
Well I’ve had 22 porphyria attacks in just over 6 years and my life consists of having a weekly infusion of haem-arginate to help minimise my symptoms and attacks. I have 50% good days and 50% bad days. I’m very good now at listening to my body and knowing when to stop and rest.
I help the British Porphyria Association spread awareness on the disease and I’ve also taken on a role as a Porphyria Ambassador in the UK with a company called ‘SNOW’ as I want to share my story and let others know that they are not alone. Life isn’t easy with porphyria and it’s really hard. We are different and we can’t always do the things we want to do.
I don’t know what’s going to happen in my future but if I can spread awareness, take part in experiments and studies and stop my sister ever experiencing the same pain and troubles I went through…. I will.
People say I’m so strong, but I’m actually continuously scared and frightened. The reason I am strong is because of my amazing support. It’s because of my partner, siblings, family, friends, my Greek family, my Medway Sport team and because of the British Porphyria Association, that I am as strong as I am. I’m honestly only as strong as my support!
Ian: a husband’s perspective of AIP
I first met Sue whilst working at a software company. As we grew closer it became evident she drove everywhere and never had a drink. Sue explained that she had acute intermittent porphyria… something I had never heard of, let alone be able to spell.
Her sister, Liz, had been incredibly ill in her teens and twenties – but Sue remained symptom free. She was always careful, steering clear of triggers, to remain healthy, and not go down the awful path her sister once did.
I couldn’t imagine life without nights enjoying a drink with my mates, but I quickly learned Sue had an incredibly strong character, and a close knit family which meant these sacrifices came and went without much fuss. Sue had a drive and determination and a zest for life that I really liked. Our life was great – late nights with friends, lots of socialising, snowboarding holidays, whilst still mindful that Sue should be sensible. She showed no signs of any issues. One particular Christmas party is where things started to change. Sue had some dodgy food – and became very ill very quickly. It took Sue a good few days to be back to her usual smiley self. A few weeks later we went on a snowboarding holiday – looking back now we know she exerted herself too much …. and soon after the signs started appearing. Speaking with Liz and their Mum, Anne, we quickly realised it was happening to Sue too.
I never thought it would happen to be honest. Sue had always been so careful. From that point I became nervous, really worried and didn’t have a clue what to expect. Despite a lot of family support, I felt quite alone – my family didn’t know much about it and certainly friends hadn’t a clue. Google can be your worst enemy in times like this, so I steered clear… all I remember thinking was that I had to stay strong for Sue, give her love and support.
Helpless is a word I’ve used to describe how I often feel with Sue’s illness. There isn’t a lot I can do to help the situation. I felt very out of my depth, knowing it would be hard to put trust in the medical profession, knowing that many had no knowledge of the condition. Hospital visits in the early stages gave me no confidence whatsoever. One night, an out-of-hours GP asked Liz what he should do as he had no idea!
Afterwards, with Sue admitted to hospital, Sue was seen by a doctor who had heard of AIP. The relief on our faces when that doctor had “heard of” haem arginate! Over the next months, we spent countless days in hospital, worried the nurses wouldn’t administer the treatment correctly, worried about a lack of suitable veins and the haem arginate being left too long. In the back of my mind, I was incredibly scared in case they did something wrong… always thinking back to what happened to Liz, how it could end up even more serious.
Not long after, Sue was made redundant from work. We’re both pretty certain her sickness record played a part in this, but there was nothing we could do. This placed extreme pressure on us financially, our lifestyle immediately changed as we had to tighten the purse strings.
Dr Penny Stein soon became Sue’s consultant and we immediately felt a sense of relief – Sue was in safe hands. Dr Stein suggested Sue could have a port-a-cath and Sue’s operation to put a port-a-cath in was a great success. A friend of mine nicknamed her “Sue-borg” which has stuck, not joking about the subject, but putting a light-hearted feel to things. My friends think Sue is amazing, with the things she has had to put up with.
More hospital visits followed, always with a worry whether the port-a-cath would be treated correctly or get an infection. But I needed to remain supportive to Sue, keep level-headed so not to increase her worry.
Homecare then taught Sue how to do her treatments at home, herself… I wanted to be involved too, and learnt the basics to help her. I hate needles, but putting them in somebody else is somehow ok! I always struggle to put the sterile gloves on – I don’t know if I get nervous, and sweaty, if I have warmer hands than usual, or what… but it’s a comedy moment, which always causes amusement. Treatments at home continued on a weekly basis (more or less).
We managed to have holidays, including camping trips, and during one of these trips away to France, we got engaged. Fast forward 18 months and we got married. Two days after the wedding, we went on an awesome honeymoon, with lots of careful planning and an additional suitcase of medications and treatment packs. For the first time, we realised how stressful travelling with medication could be. Surprisingly, getting into America was fine, we had everything we needed, including a supporting letter from Dr Stein. Las Vegas airport was a breeze. The second half of the honeymoon was Mexico… which soon became quite scary. Policemen armed with machine guns overlooking what the airport staff were doing… as Sue’s medical case was unpacked, swabbed, and x-rayed; we were extremely worried things would be taken away… or us questioned. But, we got through it. We can now look back and smile, as I don’t think we will ever feel the same – we know what to expect.
Through Liz’s experiences, we knew that Sue’s port-a-cath would need to be replaced at some point… and as the months/years went on, the treatments started to slow up. Sue was booked in to have her old port-a-cath removed, and a new one put in place. While waiting outside theatre, I remember realising that an hour had passed…. last time it hadn’t taken as long. I knew one was being removed, and another inserted so kept my head down. Another hour passed and a nurse came to see me…. I instantly knew something wasn’t right. The nurse explained that there had been complications with removing the old port. The existing line had become infused into one of her veins and they couldn’t remove it. Round two to remove the stuck port-a-cath and the operation was a success. The new port-a-cath worked well for around 3 years, where the third one was inserted under local anaesthetic because Sue was pregnant. They waited to remove the old port until baby was born, thankfully with no complications this time.
I really don’t want this to be doom and gloom, yeah its rubbish what has happened, and what Sue has to deal with, but she deals with it, and in a manner that myself and others envy. Our lives changed incredibly since Sue’s diagnosis; some friends really don’t have an idea what Sue has been through. It’s a strange illness, as you can’t “see” anything wrong with Sue, apart from the war wounds on her chest. Sue has spent years with pain most weeks, but again, nothing visibly shows…. I never know what to say or do… I want her to tell me of course, but at the same time I feel helpless, as there isn’t anything I can do to make it better.
2017 was an amazing year for us, Sue gave birth to our beautiful baby girl, Abigail. Throughout Sue’s pregnancy she was monitored closely as it can affect the porphyria. The initial morning sickness was tough and the tiring labour took its toll on Sue – but all was soon forgotten when she held our little girl in her arms. The support of Dr Stein and the clinician-led team at our hospital in Norwich was incredible, and both families too. Like all new parents, it’s been tough work, and adjusting to our new way of life took a while. I’m sure every Dad says this, but I feel like the proudest one alive, proud of Sue and Abi and what we’ve achieved.
Abi was swab tested for Porphyria soon after. We always knew it was a 50/50 chance and soon discovered that she tested positive. Sue had already geared herself up for this news, but I was simply heartbroken. I quickly came to terms with it, and can now safely talk about it without getting too upset. My perfect little princess now faces additional challenges, but I’m confident we can handle whatever is thrown at us.
In 2018, Sue started a drug trial, initially a 6-month placebo controlled study, which meant she didn’t know whether she received the trial medication or not. Only a couple of months in, Sue had a horrendous acute attack requiring hospitalisation, and a course of 4 haem arginate infusions – this left Sue very weak for some time. After the 6-month study, patients were then offered the medication which Sue began receiving. Since this time, Sue has not needed any haem arginate and is no longer having recurrent attacks! I can safely say this has been life-changing. The pain has more or less gone, the weekly infusions are no more – the last one being just over 18 months ago now. Sue is no longer in need of constant pain relief, she is physically much stronger, she actually sleeps now, is much less anxious and is looking forward to things that we didn’t think might be possible as a result of the porphyria.


Glenda’s EPP story


My name is Glenda, I am 55 and have EPP. I live in a suburb of Hobart, the capital city of Tasmania, Australia. I was finally diagnosed at 13 after many years of bizarre symptoms, doctors never could work out what was causing my symptoms. Sometimes I would get my symptoms after swimming in a pool leading to my parents thinking perhaps I was allergic to chlorine, but sometimes I had been no where near a pool. There were times I had been playing in a sandpit so my parents thought maybe I was allergic to the bleached sand but then there were times I had just been playing outside.
Looking back I really feel for my parents who had a child that would get clearly very painful, agonising symptoms (my 2 younger brothers never had symptoms after having done the same activities as me) but were at a loss as to what caused them.
Finally when I was 13 I went away on a youth camp for a long weekend. We did lots of outdoor activities including an afternoon canoeing on a very sunny day. I ended up with the worst, most agonising symptoms I had ever had. My mother took me to our local doctor but because it was a long weekend our doctor was replaced by a locum who just happened to have been to a conference the week before on Porphyria. I was sent to see the Specialist that ran the conference, was admitted to hospital for a week having lots of tests done. This also involved lots of doctors & medical students standing around my bed asking me questions which for a 13 year old was quite exciting. Finally I was diagnosed with Erythropoietic protoporphyria and we knew why I got my symptoms.
To begin with I took beta carotene capsules which had to be specially sourced but because they made me yellow I just learnt to control my condition by avoiding the sun. There were lots of times up till the 1990s when I had really painful, agonising symptoms. Since then I have avoided really bad attacks, as soon as I get any tingling which is very often I know I have to retreat indoors.
Living in sunny Australia is very challenging at times but if I want to venture outside I cover up completely as well as using hats, gloves and umbrellas. It is okay in winter being covered up but in Summer I often get hurtful comments when out & about so covered up.
I am married with 4 adult children Nathan 26, Joshua 24, Bethany 22 and Timothy 20. None of our children fortunately have EPP. I was never going to let my EPP stop me having children. My only known family connection is my 2 female second cousins who are sisters with no other siblings on my mother’s side. I remember as a teenager when my aunt first found out her 2 grand daughter’s had EPP she thought the world had ended because she had witnessed me as a child before I was diagnosed sitting on her porch in Queensland trying to get a cooling breeze with my hands, feet, face draped in cooling wet cloths. Something is much easier to live with when you know the cause.
My husband & I love travelling, I just have to cover up and cope with hurtful looks & comments. We really love cruising. I find cruises really liberating because during the day I can walk around inside the ship in my summery clothes and sandals participating in all the great activities and then at night after the sun has set enjoy the freedom of being up on deck.
I often refer to myself as a vampire which upsets my husband, he prefers me to call myself a ‘shadow walker’.
I have never met another person with EPP. I fly to Melbourne once a year to be monitored by Melbourne Hospital’s Porphyria Clinic. It is so refreshing to be treated by doctors that truly understand what it is like to have EPP, when I first met these doctors they said ‘no one really gets what it is like for you having EPP do they.’ Many times over the years I have had to do my own medical research.
It is actually our love of cruising that now finds us in self isolation due to the fact we disembarked a ship that has since had a passenger test positive to Corona Virus. This was one of many cruises we have done and once we all come through to the other side of this pandemic we will again go cruising.
I hope you have found my EPP experience from the other side of the world interesting.
Natassja shares her experience of being a Mum to children with EPP



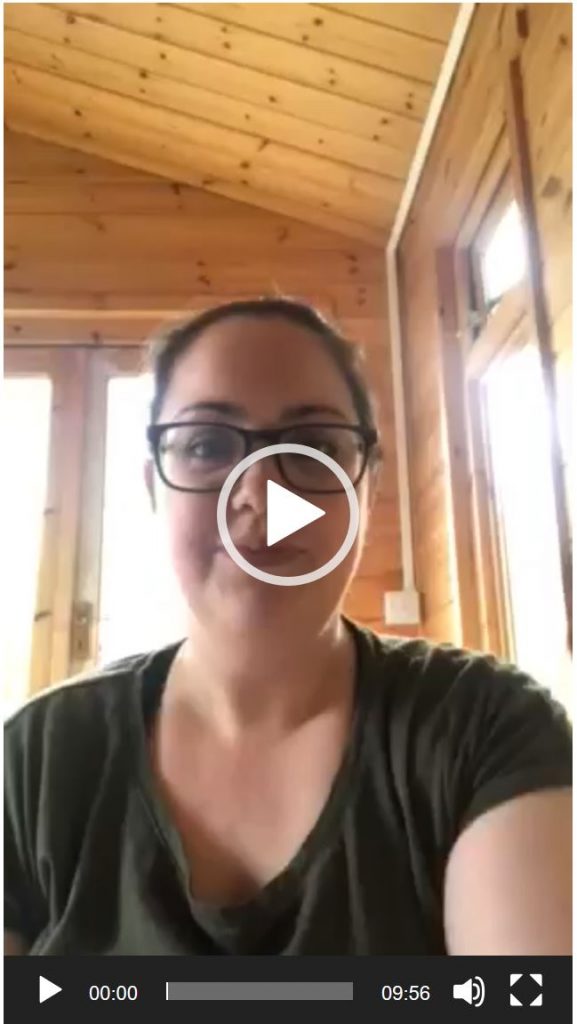 Natassja’s candid account of the life adaptations and the psychological effects of having children with EPP will resound with many other parents.
Natassja’s candid account of the life adaptations and the psychological effects of having children with EPP will resound with many other parents.
This is a must watch video.
Maddy and Colette share Maddy’s EPP journey
Maddy’s story
My name is Maddy. I am 13 years old. I have EPP.
This is what EPP feels like for me:
At first it tingles, then it starts itching, next comes the burning, like it’s on fire. Then the swelling and my skin goes tight.
Nothing much helps the pain. My mum has given me antihistamines and used a cold pack. I have taken cold showers/baths. But it doesn’t really work for me.
I just have to make sure I get inside as soon as possible in a dark room. I take paracetamol for the pain. The tingling can go but then it wakes me up again in pain.
It really hurts and can take a long time to go away. Then the swelling comes. It is usually my hands but when it’s really bad, my full face too.
I am lucky that I mostly only get it during the summer months, but I have to be careful on sunny days in the spring too. Sometimes I have a really bad swelling in the summer, but I can be in the sun longer towards the end of summer.
I have the proper sun cream now and make sure I wear a hat and keep covered. I have my curtains closed at home and I don’t sit near windows in class. I saw a doctor who talked to me about everything and said there are lots of things I can try. I am going to try Beta carotene and there are other treatments as I get older.
I do get sick of all the blood tests and was really sad when my mum said I couldn’t go to Spain on my school trip.
I have been asked if I’m a vampire but mostly everyone has been really kind, just wanting to know about EPP.
I am okay 😊 I still go out with my friends and have fun.
Written by Maddy’s Mum, Colette
My daughter Maddy was finally diagnosed last year with EPP at the age of 12 This was after years of wondering what was wrong. She has two siblings but the only one with EPP.
The earliest memory I have of Maddy showing signs are age four. We had gone to the park after nursery and she suddenly started screaming saying her hands were hurting. I couldn’t understand as she had tons of sun cream on but reapplied on her hands anyway. This didn’t help, we went home as she didn’t settle. The doctor thought it was prickly heat and prescribed antihistamine medicine. Over the following years I kept ensuring Maddy took her antihistamines and was always covered in suncream. This didn’t stop the swelling though or the times she would wake up screaming during the night saying her hands felt they were on fire. She had cool baths but none of this helped. I have taken her to A and E but the swelling had gone back down by the time she was seen. Back at the doctors they thought it could be an allergy so referred her for testing. I could not understand as this only happened in the Summer.
It was time for Maddy’s school residential. She was going away camping for 2 days to do adventure activities. I was apprehensive as it was summer so ensured she had suncream, hat and plenty of antihistamines. I saw the photos online of her joining in with the water sports. Then later that afternoon her teacher called and said Maddy was upset.They were fantastic making sure she had her antihistamines and suncream applied. I spoke to Maddy also thinking she was home sick too ( I feel really guilty about this now) She didn’t have swelling at the time. The next thing was a call to say Maddy was in hospital as her face had swollen. This had never happened before, it was always just her hands. I was beside myself as didn’t know what was wrong. I knew at this point it was definitely not prickly heat!. When I saw my little girl my heart broke. The picture is taken from the hospital. She had a red band on and the doctor said she had suffered from an allergic reaction. They did a finger prick test but didn’t take bloods. They prescribed antihistamines again! My head was spinning and I just knew that it wasn’t right. I took her to her own GP that day so he could see. He referred her back to the skin specialist for more testing.
Her swelling took days to go back down and her skin was so tight and dry afterwards.)In the meantime my sister had looked up her symptoms and rang me to say that Maddy had all the signs of a condition called Erythropoietic protoporphyria . As soon as I looked, I knew. I spoke to my GP who looked it up and also thought Maddy showed a lot of the signs. He referred her for the specialist test via her skin specialist. Getting the result was a bit of relief to be honest as I finally knew what was wrong with my beautiful girl, but also felt extreme sadness that it had taken this long and for what the future would hold. I feel so proud of how Maddy has dealt with this. She said she won’t let it stop her from achieving her goals in life. We have now seen a fantastic specialist in Leeds who Maddy will see every year from now on. She explained everything and tested Maddy for her vitamin D levels. She is now on a super high dose and finally has the correct suncream. She was really positive and said although this is lifelong there are treatments available that help some people. Maddy will be starting Beta Carotene soon and we will look at other options as she gets older.
We do have some challenging days like when I decided that it was best for Maddy not to go to Spain on her school trip for now ( I just wouldn’t settle with worry,).
But Maddy remains positive and even though we still worry about summer. We now know what we are dealing with, what to avoid and I ensure Maddy has all the correct protection in place now.
Attached are photos of Maddy in hospital and one of what she usually looks like.


Samantha shares her story about EPP

Hi, My name is Samantha, I first knew I was a little different when I was 4 years old. I was on a camping trip with my parents. It was really sunny and I started to itch like mad, then I started crying and scratching my arms to pieces. My mum rush me to A&E and they said it was likely sun stroke as I had no other reaction and I otherwise looked fine. They told my mum to keep me out of the sun. The next year a similar thing happened. This time my mum took me to the local GP and asked what this was as it was the second time this had happened. The thing about my porphyria is that is doesn’t always show external signs. So I had nothing other than itching and pain.
This continued until I was 8 years old. My friend asked me to go to the beach with her, my mum explained that I was sensitive to the sun and I wouldn’t be able to go out in the sun much. 8 hours later I returned and my hands and feet were so swollen I could barely move. The parents of my friend were really apologetic not realising what their “day of fun” could have done to me. Again we went to A&E, I was told Sun stroke, but this time they told my mum she was attention seeking and to just take me home and put me in a cold bath. When I got home, she did as she was told. As soon as I went into the bath I screamed so loudly that a neighbour (a paramedic) rushed around and asked if we were ok. My mum explained and he said, that doesn’t sound like sun stroke.
For years we went back to the doctors, I was given creams and antihistamines, but they kept saying there was nothing wrong. They also accused me of being a hypochondriac and even threatened my mum with social services… thinking she was making it up for attention as well. I have no idea why we were treated so horrendously, but we were. As the years went by me and my mum realised that it seemed to get worse when I was out in the sun, especially on hot days. My mum also recognised that during an attack I would be cold to touch and that her touch would make me scream because the heat of her body was painful to me. We considered whether it was an internal thing to do with heat. But not one doctor would look into it.
When I was 16 we moved to Milton Keynes. We spoke to a new doctor and she tested me for Raynauds Syndrome. It wasn’t what I had but at least I was being taken seriously. However no more tests were done. When I was 18 I moved down south for work. My doctor was a dermatologist, so I booked a double appointment to go in and speak to her. She said to come in if I had a reaction. So as soon as I had a reaction I went back to her, she couldn’t see anything and admitted she did not know what it was. So the mystery continued. I moved around a bit more for work and ended up back in Milton Keynes with my mum. I got married and had 3 beautiful children. This however caused me to have some other health issues. As part of those I was prescribed Amitriptyline. In the first few days I felt very dizzy and I could not find the leaflet, so I went onto the NHS website and looked for the leaflet for Amitriptyline. On that leaflet it said that dizziness can be a normal side effect, but it also said not to take if you have Porphyria. I am naturally a curious person, so I researched what Porphyria was. It said that there were Acute and Cutaneous Porphyrias.
After spending years trying to find out what my condition was I knew what cutaneous meant so I read on. As soon as I read the symptoms of Erythropoietic Protoporphyria I knew what I had. It was exactly what happened to me. I also itch a lot and we never really noticed before. It was just something I have always done. So I went to my GP and I said after 28 years this is what I have! He said it was highly unlikely I have this but he would refer me to dermatology. A few weeks later I went to dermatology and the lady doctor said it was definitely not EPP, I said I am sure it is. I would not leave so she gave in and let me have the tests done. She was so sure I did not have it that she discharged me back to my GP before the tests were even completed. 6 weeks later I got a phone call asking me to come into see the head of dermatology the following Friday. Needless to say, at the age of 32, a few weeks before turning 33, I finally had my diagnosis of Erythropoietic Protoporphyria.
I am still struggling to find a specialist I can get to that knows about this condition, but to be honest, I am not too worried. I know what helps this condtion, but now I know even more and I haven’t had an attack in 4 years now. It is hard having an invisible condition, even harder when it is a rare one. But I am happy I now know what it is. It has brought me an understanding that I never believed to be possible and on top of that I have found that my kids are unlikely to inherit it. This, for me, was the best news ever as I did not want them to suffer like I did. I will let them know about my rare condition though as they carry a faulty gene and will need to know for their future children. But they have a choice and they have knowledge.
Thanks for reading my Porphyria Story!
Samantha
Life with EPP by Victoria
My name is Victoria and I live in Mansfield, Nottinghamshire. I’m 35 and I was diagnosed with EPP at the age of 24.
For most people, the long, sunny days of summer are something to be savoured, with barbecues, trips to the beach and lazy afternoons in the park are what everyone waits all winter for.
But for me I dread the coming of the summer months because I suffer from an extremely rare genetic condition called erythropoietic porphyria (EPP) which means I am allergic to sunlight.
I was finally diagnosed after a lifetime of doctors not being able to explain why I felt incredible pain whenever I went out in the sunshine and then suffered from terrible swelling and blistering afterwards.
The way I would describe the pain, is that it is like having 200 needles stuck in your skin but about a hundred times worse, it feels like a really intense burning.
The doctors thought at first that I was making it up and it was all in my mind, but I was admitted to hospital every time I went on holiday with severe swelling, blisters and scabs. My medical history states anaphylactic shock, depression, self-harm and this was never the case.
I have always had reactions to sunlight but it was never picked up on because the condition is so rare and nobody else in my family is affected. When I was diagnosed I travelled to see a specialist in Cardiff who explained more about the condition.
EPP is caused by a faulty gene that causes a build-up of a porphyrin chemical in the blood and leads to sensitivity to sunlight (also known as a phototoxic reaction). It also affects the production of haemoglobin which leads to a deficiency in iron and can cause anaemia, as liver problems can also arise, I have a liver test every year too.
There is no cure for EPP so it has to be managed by avoiding being out in the sun and covering up from head to toe whenever you have to go out.
I am constantly having to think about the weather and I have to make sure that if I am planning to be outside for any period of the day that I have somewhere indoors that I can retreat to.
I also cover up completely before going out — putting on a hat, coat and gloves and I wear reflective Dundee cream to minimise the risk of a reaction. Normal sun cream is ineffective because it only blocks UV light and not visible light.
I miss not being able to sit in the sun but I have to be so careful. I have never been abroad as it puts me off because I can’t use my hands or anything if I have a bad reaction.
Over the years I have learned that I need to be open and honest with those around me, if I don’t then how can I expect them to understand me.
I am very fortunate to have amazing parents and some absolutely fabulous friends; they all help me get through. Lean on those around you, don’t be afraid for they are there with you.
There are times where I have to say no to the nice days out and it does upset me. But, as I said I have some really good friends and we organise doing things that we can do inside or where there is somewhere inside to go. My friends will set up places for me to sit in the shade if we are outside, I can cover up around them and they don’t make a massive deal of me being different, it makes me emotional on occasions because they make me feel so special and part of them.
Hopefully one day I will be able to enjoy the sun but for now I have to keep strong and support my porphyria community, a way that I do this is by running a support group on facebook called ‘Erythropoietic Protoporphyria Support Group’, I run this with two other admins who are amazing, I am proud of this group because it does exactly what it says on the tin just like the BPA and APF.
My spirituality helps me stay strong when I feel down and I love to sing as it helps me escape.
When I go out covered up I often get strange looks from people and sometimes it hits hard, I was bullied at school because of my condition, so I am keen to raise awareness about EPP so that people are more understanding.
I want people to know, because I have had occasions where others have taken the mickey because they don’t understand.
I also want to keep raising awareness because there may be more people out there struggling, who may have the condition and are being told there’s nothing wrong with them!
A new drug was approved by the EMA a few years ago for the treatment of EPP, but this is not currently available to patients in the UK on the NHS. The treatment, which is an implant under the skin, helps patients to become more resistant to light and is being used in various countries around the world. I have previously had light therapy which aims to do a similar thing, but unfortunately it didn’t work for me, I have heard stories from others that is very successful for them.



Paula’s PCT journey


My name is Paula and I started my porphyria cutanea tarda (PCT) journey as a healthy 49 year old who had never been seriously ill, broken a bone and had never been in hospital.
In 2015 I had started to experience extreme fatigue and trauma to my hands which I had put down to 60 hour weeks at work and an old pine bed base which would strip the skin off my knuckles every time I tucked the sheet under the mattress. After one of my colleagues accused me of being in a bare knuckle fight, I bought a set of heavy duty rubber gloves to combat this and never gave it another thought.
In June 2016 I went on holiday to Tenerife and on the return flight noticed that I had some blisters on my knuckles. Ah, I thought this is the reason for the skin peeling off my hands and I booked an appointment at the doctors thinking I perhaps had an allergy to washing powder. The doctor and I swapped notes and I sent him my photographs of the blisters and scars from the previous skin traumas and fortunately for me he was a junior doctor that was being assessed, so he took lots of notes and confirmed with me at the end of the consultation exactly what I had said to him. He then referred the notes and photographs to a dermatology unit as that way I would get a faster diagnosis rather than having to wait for an appointment.
He telephoned me a few days later asking me to call into the surgery for an envelope which included a blood test referral and a urine sample bottle with specific instructions to follow for both. I noticed that he had written on the paperwork to test for porphyria, I didn’t know anything about this so I made the mistake of Googling it! I then spent the next hour crying and was in deep shock. Being a positive person, I decided that of course the results would come back negative. After completing the urine sample in the dark and the blood test which was initially refused at the doctors due to the fact that they hadn’t got the specialist medical equipment necessary, I was referred to the local hospital for the blood tests where they wrapped a kitchen towel around the test vial. These were all sent off for analysis and the results came back positive. My ferritin levels at this point were around 400.
Again, I was floored. My doctor gave me the standard PCT leaflet and referred me to a dermatologist unit. That was all the information I was offered. I booked to see a private dermatologist (£150 later) who told me not to go on a sunny holiday, not to drink any alcohol and said that I couldn’t take the oxytetracycline tablets that had been prescribed for Roseacea 5 years previously. Now I was deeply distressed as it was my 50th birthday in a couple of months’ time and I was going away with family and friends to Cuba on an All Inclusive holiday. The dermatologist advised me against going but everyone had already paid and she waved me out the door with a hearty “have a lovely dry Christmas” and a big smile on her face. Was this all such a big joke? It appeared that it was and that I was just missing the funny side.
My appointment came through for the NHS dermatologist and so I went along fully informed with the information that I had got from my doctor and Google. Only to be treated with distain and told that I couldn’t have any treatment at all as the two options available were too dangerous. Again the standard PCT leaflet was offered to me by the dermatologist which showed the two treatments of which I could have neither. One was anti-malaria tablets and the other venesection. I felt as though I was being fobbed off, I was not being given any further information on PCT, it appeared that nothing was up for discussion and I was made to feel as though it was all my fault that I had the disease.
I got in touch with Sue around this time who offered me an amazing amount of personal support and also went back to my doctors to complain about the lack of treatment. While my doctor got the ball rolling again, I turned to Google. I went to an online chemist where I requested the anti-malaria tablets as I was going to the Dominican Republic for 6 months and checked on the “give blood” website to find out if I could give blood. There was nothing mentioned on the website about PCT so I signed up and went along.
2017 flew by and I was still giving blood and taking the anti-malaria tablets when the dermatologist saw me for my 6 months check up and said that they would refer me for venesection as “I had done a really good job of getting my ferritin levels under control”. My ferritin levels were around 180… ok….
I was referred to Heartlands Hospital where I had three venesections over the next three weeks and this bought my levels down to under 50 which I was advised that this was what they should be. I read an article shortly after this where hemochromatosis patients could go and give blood on a monthly basis. So I enquired about this as it would be easier for me to give blood at a convenient time and also that they would be able to use the blood. When I told the assistant that I had PCT she immediately struck me off, when I enquired why she said “well you can acquire it from advanced liver disease or hepatitis C or HIV”. All news to me, so I responded with “ok so you think I am an alcoholic drug taker then” … silence ensued. Well you know she wasn’t actually saying that… but the penny started to drop as to why people were treating me with such distain.
Where am I today…
The dermatology unit discharged me as there was nothing further that they could do – thank goodness for that – it was wasting everyone’s time! I am still being monitored by the Haematology department in Heartlands Hospital who are lovely people, they don’t judge and look after all the cancer patients as well as PCT’s and hemochromatosis patients. I still don’t know to this day how I have ended up with PCT – no one in the family has it, I am not an alcoholic, I don’t have HIV or Hepatitis C. However, I was taking oxytetracycline which is a potential trigger, I am also menopausal so a drop in oestrogen could have also been a trigger. Either way, I have it now and have to deal with it. I still go on holiday in the sunshine, I don’t cover up as I do tan but I use a high sun protection factor and stay in the shade, I eat meat rarely and enjoy a glass of wine or a gin and tonic occasionally. I exercise on a regular basis and cut my working hours down to reduce stress. The skin trauma to my hands has stopped and the condition of my skin has improved, I will obviously have to live with the scarring.
I am enjoying my life as it is to be lived and not dictated to by those who read a leaflet. I am also relatively comfortable with not having the answers to the “why me” question as it would appear that no-one has probably got that answer.
I appreciate all the support that I have received from Sue and Liz at the BPA and also the support of you all via Facebook.
A life with VP, by Leigh
My name is Leigh, thank you for inviting me to share my experience of living with variegate porphyria (VP) for the past 31 years.
Its pronounced Porfiria, it is very rare, it is genetic, you cannot catch it! The gene for the South African form of variegate porphyria was introduced into South Africa in 1688. Although there are 9 mutations, on diagnosis of mine, it was found the R59W gene was defective. I have 3 daughters, the eldest has the same gene defect as does only 1 of my twins. (They were tested in the UK back in 1997 in Cardiff). There is NO cure.
MY own personal opinion is that the more you know about your own type, the more you are confident to manage it on your own, because trust me you will be on your own with this, even in hospital. It has taken 23 years now and only just have I been fortunate to be assigned a consultant in Leeds who has also worked together with my GP to put a hospital plan together on my NHS patient record so that I need never go through my experience of June 2019 again, and any further attacks and admissions may be handled better.
I found in my 20s and early 30s that the abdominal pains played a major part in discomfort and pain. I can only describe the pain as though someone was burning you on both sides of your body but underneath the skin. For years this seemed to settle and when I felt an attack approaching, I mixed my own solution of glucose powder and you can drink as much of that as you like. I also have outgrown, or something has changed, but I do not have the focal epilepsy (right-hand side) anymore.
I have been having stomach issues for the past 5 months, and they put me on a liquid diet carefully of course due to the porphyria and then introduced me back on solids. I do find that by having probiotics and playing about with my diet in recent times to have helped me to get it to a more comfortable level. My iron is low, my immune system is low, I have now been told I also have an underactive thyroid and all these things cannot just be treated with medicine with the porphyria. I am sure the sport models, like myself, will find they will also NOT give you HRT.
My biggest concern is when something ELSE is not quite right, and I am sorry if I upset any professionals but a lot is blamed on the porphyria unnecessarily. I walked around with heart monitors, machines, sending tracings down phones, being out of breath, couldn’t lie on my left side, still can’t and have just been struggling for the best part of 6 years. They even told me it was anxiety. Finally, when someone would listen, he suggested a scan they found I had a mitral regurgitation. The valve gets scanned every 3 years and fortunately in my own case, it is not getting larger and no surgery is required.
Skin is another story. The skin on my hands and arms are very fragile. Most scrapes or bumps lead to the skin being damaged and a very lengthy process and time for it to scab and heal, whilst watching that it does not get infected. When the blisters form they get this liquid in them. I try break mine otherwise they get bigger and bigger and I find the liquid that comes out really burns. These leave scars, again take a long time to heal. For the young ones with VP, I can say it has got better with age, more manageable and I just cope with it all so much better, so there is one joy of getting older with it! I also get clumps of white dots under the skin which my consultant says is milia.
SUNBLOCK!
Those with VP are extremely sensitive to sunlight. I can never find Dundee cream so I use a 50% factor EVERYDAY. Growing up and living in South Africa for the most part of 31 years, I was from the generation that did not know or wear sun block. Please, please, do take special care. I spent 3 years having 4 weeks of, then 6 weeks of chemotherapy to then find I then required 5 lots of PDT (Photo Dynamic Therapy) which is extremely painful. Treatment only takes 10 minutes each time, but I cried like a baby in sessions 3, 4 and 5.
Even the professionals are not always in agreement on how to treat you when symptomatic. I have yet to still learn if haem arginate would be suitable, which one consultant would prefer; however, back in 2011, in Sheffield, they said absolutely not with VP. I always find this disease should have been called ‘confused.com’.
BUT, we are fortunate to be here and I am very grateful and have learnt ACCEPTANCE. It does get better with age! And the joke why did you come to the UK – well uhm for the weather…. It’s great for me😉
Hope you have found my own experience with VP enlightening and from myself and my family thank you BPA for raising awareness.
Pam’s experience with HCP

I have Hereditary Coproporphyria (HCP)
Before I was diagnosed I had many years of being in excruciating pain, sensitive skin, blisters on my hands and being very unwell. Many visits to the hospital and being told there was nothing wrong with me. Throughout my pregnancy, during childbirth and following a hysterectomy at 24 I fell seriously ill with life threatening conditions. Of course at the time we didn’t know it was Porphyria, it was a horrible time.
In 1995 I visited a dermatologist, I had blisters all over my hands and he thought it was a condition called Porphyria. I had blood tests and these were sent off to Cardiff to Professor Elder and I was diagnosed with Hereditary Coproporphyria (HCP). What a relief, I cried, I finally found someone who believed me!!!!
There was no medical list of what medications I could or couldn’t take and it was a very difficult and stressful time trying to find any information on my condition. I read about a Dr Moore in Scotland and after some research I found the number and rang it. Unfortunately he had moved overseas, but luckily his ex-secretary picked up the phone and kindly sent me the last booklet he wrote. I was so delighted and it became my bible.
In 1996, still trying to find as much as I could about porphyria (which was very little) and still struggling with attacks, I found Professor Tim Cox, who had moved to Addenbrookes Hospital. He was my life saver, finally someone who understood and was helping me.
My family were tested and my father, brother and one of my nieces were diagnosed with HCP, my daughter was negative. My father felt so bad that he had passed it on, but he didn’t know he had it. My father and brother didn’t experience any attacks but my niece, in later years through her 20s, had a few attacks.
In 1998 Professor Cox organised a meeting in London with a few colleagues from different parts of the country who specialised in, or who had a keen interest in Porphyria, along with a few patients, which I was one. The meeting was to help to see if we could start a support group ran by patients with assistance from a medical team. The BPA was formed, starting with a few people you could talk to who understood my experiences. It was amazing, we didn’t feel so alone and others would at least have a point of contact. The hard work began and is continued by some amazing people that have brought the BPA to where it is now and where it’s going to be in the future.
Years’ later my brother had open heart surgery and trialled new drugs for his heart condition, he also has diabetes but neither of these caused any HCP attacks. I have a few medical conditions, B12 deficiency, Malrotation of small and large bowel with 50% mid line shift, osteoporosis, cardiac arrhythmia and glaucoma. Some of my health conditions trigger the Porphyria and vice versa, it can be quite a balancing act at times, with many times on blue light to the hospital.
Menstrual cycle (later the menopause), stress and infections definitely trigger attacks for me, I find that I’m not sick during attacks, but suffer with severe pain in my abdomen and legs, back aches, feel weak, confused (only had loss of memory twice) and my skin becomes very sensitive, sometimes blistering. In the early years my attacks would definitely be more frequent, I felt like I was just getting over one and another one started. I was told by a Porphyria specialist that people with HCP usually come out of attacks and recover quicker that those with AIP, although the evidence is not gathered, hopefully in the future we will know.
I manage my attacks in various ways, medication, sleep, rest, eating regularly and using breathing and meditation techniques. Over the years I have found that the breathing and mediation techniques have helped me tremendously and I have developed these skills to help manage the pain and reduce the recovery time of the attacks or have stopped them escalating. I’ve used these techniques a number of times for medical procedures, like urethra dilation, very successfully.
I’ve not allowed the Porphyria to affect what I want to do. When traveling overseas , I research and plan in advance, ensuring I’ve got all my medical information, i.e.; last bloods results from my GP, ensure my medical alert details are up to date, my emergency treatment guidelines and the nearest hospital to the accommodation.
Professor Tim Cox has retired (a very sad day for me) and I am now under the care of Dr Penny Stein who is fantastic, very caring and always responds very quickly to any questions I ask, and the support of my GP. I am very lucky to have had and continue to have great support from these great medical professionals. I don’t have many big attacks now only a few a year. I have small ones (mild symptoms) and if I start to feel unwell I act straight away and start my routine, of rest, sleep, eat well and meditation.
Lianne’s HCP story


I’m 45, married with two children, and was given my diagnosis of HCP at the age of 21. The first acute attack that left me hospitalised was in April 1996; I was admitted with acute abdominal pains, but after investigations found no apparent cause so I was sent home. The abdominal pains continued and I began having prolonged convulsions throughout each day. Again, being admitted to hospital (a week after being discharged) nothing was diagnosed and I was discharged again. The abdominal pains worsened, accompanied by nausea and constipation and convulsions lasting for up to 5 minutes at a time.
At this point I had severe muscle weakness and had increasing difficulty walking, not only that, but my speech began to slur and stutter. My speech and mobility continued to worsen, so my parents at this stage brought me home from university and took me to my GP. After checking in my eyes and doing other examinations I was admitted to hospital with a suspected brain tumour. CT scans revealed this was not the case, but I was now completely paralysed and could only stutter a few words. I had every test and scan imaginable, but was told it all must be stress related to me being a student and holding down several part-time jobs. They stopped the medication I was taking (mefenamic acid for bad period pains) and over the coming weeks my speech returned and with physio I learnt to walk again.
I returned to university, although my health had its ups and down, mainly my limbs being jerky and speech problems. If the abdominal pains got bad and I had convulsions, I would suffer at home as I’d been told it was stress. Nearly 7 months later I received a phone call from the hospital asking me to come in as some mislaid blood tests had been found from when I was admitted in April. When I arrived at the hospital, in not so good health, I was admitted and spent weeks having neurological tests done. I was asked if anyone in my family had Porphyria. I’d never heard of it. My urine was sent to Cardiff for analysis to determine what type I had. It was a few weeks until I was told it was Hereditary Coproporphyria (HCP), but I remember the relief at being told what it was. I finally realised I wasn’t going mad and everything began to make sense. I was also told that mefenamic acid was the trigger.
Due to the nerve damage from so many attacks it took until 2003 before my nerve conduction tests showed a complete recovery. However, the nerves in my arms and hands were very poor, so in 2004 I had carpal tunnel surgery on both hands. My porphyria settled down for a few years and only a handful of hospital admissions took place between 2004 and 2011, for which I was treated with glucose drips and sent home.
A lot of the time I would have attacks (in line with my menstrual cycle) but having a young family I would not go into hospital. I’d meet with Dr Badminton a few times a year and he advised my GP to look into other possible health problems that could be the trigger for my attacks, as I have always stuck to my safe list of drugs and have avoided alcohol since being diagnosed. In January 2011, I was referred to a gynaecologist as my period had not stopped for 3 months and was found to have a fibroid (1.5cm) and this could be the cause of the attacks. It was recommended I have hormone treatment to suppress my ovaries, but due to my GP not informing me of this I continued to have monthly attacks, by January 2012 they had got so bad that I agreed to stay in hospital. Tests revealed my fibroid was now 10cm (and growing rapidly) and I also had endometriosis; they began Prostap injections to try to control my hormones and I was told I would need a total hysterectomy including removing my ovaries, they operated in April 2012, putting me into a medically induced menopause at the age of 37.
In 2012 I spent 7 months in hospital as my porphyrin levels were so high and would have a new central line fitted each month to enable me to have haem arginate infusions. Before I could be discharged from hospital I had to do the steps test. Having so many repeated attacks my mobility was affected (although not complete paralysis like in 1996) and I had to show the physio I could manage to walk up at least 7 stairs.
I was fitted with my first portacath in September 2012. This allowed me to be at home and have weekly infusions as an outpatient before moving to homecare in 2013. Unfortunately, my first portacath procedure was not a good experience. The surgery was training a radiologist to fit portacaths and for some unknown reason attached the portacath to the outside of my skin (which is why I have a photo of what it looks like), it was later put under my skin after I pointed out the error! The photo became a useful training tool for nurses if they are learning to access my portacath.
Eight years on from having my hysterectomy and beginning regular haem arginate infusions my life has changed dramatically. Every day is different and managing pain is key. I have suffered other health problems since 2012, having a minor stroke which led to them discovering I have AV block (heart condition) as well as being diagnosed with osteoporosis. Having other health issues to contend with it is sometimes difficult to distinguish which pains and symptoms are porphyria related and too often the porphyria is the one I don’t give much focus to. I have learnt to live with a chronic illness by not allowing it to control me. It’s no longer my first thought when I wake up, but there are times in the day or week when I will feel really nauseous or fatigued, or it could be abdominal pains and it’s like porphyria is reminding me – ‘I’m still here!’
One thing throughout this journey that I want to share is that you should set yourself a challenge. In 2015 when I recovered from my horrific year in 2012 I decided I wanted to achieve a lifelong ambition – to run the London Marathon. My family and friends thought I was crazy. I’d put on 5 stone in weight and hadn’t exercised since 2011 but I gave myself 6 months to make it to the start line. In October 2015 I began running (shuffling more like) a couple of times a week and in April 2016 I completed the London Marathon. It wasn’t easy and it wasn’t fast (6 hours) but I did it. My nurse came to do my infusion the next day (picture included), and it did take a while to recover. I don’t often share life with porphyria on social media but on this occasion I did to try and inspire others to achieve their goals.
I feel very lucky to have such a good team of haematologists who have taken a keen interest in Porphyria and liaise well with Dr Badminton to ensure I get the correct support and treatment, and I’m not afraid to go into hospital at the first sign of an attack. I have a fourteen-year-old daughter who has inherited HCP, but I hope and pray that she never has an acute attack as my twin sister, elder brother and mother (all with HCP) have been fortunate never to have suffered.
When you live with a rare illness and become very sick with it, your family have to adapt to changes as well as yourself.
Knowledge is key, and more often than not we, as patients, are the first point of reference for information about Porphyria. I always remember to carry a safe list of drugs wherever I go!
So many of you also got involved in our bRED campaign during the awareness week itself.
Thank you for your amazing input, your imagination and your dedication to making the week such a great one for porphyria awareness.
